
Qu'est ce que la maladie de Von Recklinghausen ?
C'est une affection génétique, longtemps confondue avec la "maladie d'éléphant man". Elle atteint 15 000 personnes en France.
Les maladies génétiques :
Les maladies génétiques désignent l'ensemble des maladies qui sont causées par un ou plusieurs gènes défectueux ou par une anomalie chromosomique. Elles peuvent être héréditaires ou non. Ainsi, les maladies géniques qui concernent des gènes défectueux sont transmissibles à la descendance. À la différence des maladies chromosomiques qui ne sont pas héréditaires. La NF1 fait partie des maladies génétiques rares les plus connues, en France elle touche une personne sur 3000 à 4000.
Le blog que nous avons créé vous informe sur cette maladie rare et vous dirige sur le site de l'association Kokcinelo
Le chromozome 17 :
Le gène responsable de cette maladie est situé sur le bras long du chromozome 17. Il fabrique une protéine , la neurofibromine, responsable de la maladie. La neurofibromatose est une maladie plus ou moins sévère (de type 1 ou 2) avec une évolution imprévisible ce qui empêche d'effectuer un pronostic précis.


Les symptômes :
Les symptômes sont présents dès le plus jeune âge, on observe :
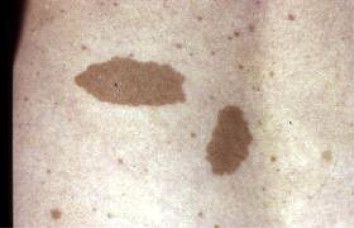
► des marques brunes claires sur la peau, appelées taches « café au lait » d'un diamètre supérieur à 1,5 cm après la puberté ou à 0,5 cm avant la puberté
► des taches de rousseur sous l'aine ou sous les bras
► des tumeurs non cancéreuses sous la peau (ou plus profondes dans le corps)
► des taches brunes sur l'iris, appelées nodules de Lisch
► des annomalies osseuses, comme les jambes arquées, une petite taille, ainsi qu'une scoliose
► des tumeurs sur le nerf optique (gliome)
Quelques symptômes en image :





Diagnostic :
Avant pour que les médecins puissent commencer à diagnostiquer la maladie, il fallait que le patient présente au moins six tâches café au lait d’un diamètre supérieur à 1.5 cm sur la totalité du corps. Après la confirmation de ce symptôme, on pratiquait une IRM afin d’observer des bright spots dans le cerveau. Ensuite on procédait à une observation du profil de l’œil à l’aide d’une lampe à fente à la recherche d'hamartome iridien.
Si tous ces symptômes étaient réunis on réalisait un séquençage du gène NF1, et dans 80% à 90% des cas le gène présentait une mutation.
Actuellement des techniques permettent d’améliorer le séquençage des gènes, on procède donc au séquençage après le premier diagnostic. Chez les enfants de moins de cinq ans, des visites régulières chez un ophtalmologiste sont obligatoires afin de détecter un possible gliome du nerf optique. Les adultes quant à eux s’auto-palpent afin de dépister eux même le gliome.
Complications :
En cas de complications les tumeurs peuvent devenir malignes, il se développe également des tumeurs cérébrales, des tumeurs des glandes surrénales, des tumeurs localisées en bas des reins. La première peut entraîner des déficiences mentales, des crises d'épilépsies et des troubles de l'apprentissage et du comportement. On différencie les tumeurs cérébrales des patients atteints de la NF1 aux tumeurs des autres patients qui nécessitent des thérapies différentes.
Les tumeurs des glandes surrénales entrainent une augmentation du taux d'adrénaline dans le sang et donc de l'hypertension.
Ces complications sont imprévisibles d'ou la difficulté de donner un pronostic au patient.
Dépistage :
Il est possible de dépister la maladie dans deux cas :
Lors d'un diagnostic prénatal ou préimplantatoir ou lorsque l'on envisage un diagnostic précoce chez l’enfant présentant des manifestations incomplètes. Ces techniques sont pratiquées dans trois centres en France: le CHR d'Orléans, Hôpital Antoine-Béclère, Hôpital Necker.
Le séquençage de l'ADN est une technique qui a révolutionné la biologie moléculaire dans les années 1970. L'ADN contenu dans chacune de nos cellules, contient l'information nécessaire à l'expression des gènes.
La méthode de Sanger, qui repose sur la biologie moléculaire, est désormais la plus utilisée. Bien que le séquençage ait beaucoup évolué et soit désormais automatisé, il repose généralement sur l'utilisation de composants biologiques qui existent naturellement dans les cellules.
Pour le séquençage, tout se passe initialement dans un tube, en présence de l'ADN à séquencer, des nucléotides normaux et des nucléotides modifiés, d'une amorce et de l'ADN polymérase.
L'ADN polymérase utilise aléatoirement les nucléotides présents dans le tube pour copier le brien mère en synthétisant un ADN de séquence complémentaire, du moment que la base (A, C, G ou T) est respectée, en respectant la complémentaritédes bases A-T et C-G.
Lorsque l'ADN polymérase choisit par hasard un nucléotide modifié (didésoxynucléotide, sans groupement -OH) pour synthétiser le brien d'ADN complémentaire, la synthèse s'interrompt prématurément. Chaque didésoxynucléotide étant associé à un fluorochrome différent (A vert, T rouge, C bleu et G jaune) une chaîne qui se termine par exemple par un A sera verte.
Puisqu'un grand nombre de réactions de synthèse ont lieu dans le tube, il existe des fragements de différentes tailles (correspondant à un arrêt de la synthèse à chaque didésoxynucléotide) et de nombreuses copies d'une même taille. Ces chaînes commencent toutes au même endroit sur l'ADN à séquencer (déterminé par la séquence de l'amorce utilisée), toutes celles qui possèdent la même longueur se terminent donc par le même didésoxynucléotide marqué.
Il est alors possible de séparer les chaînes d'ADN obtenues en fonction de leurs tailles, sur un gel d'acrylamide en présence d'un courant électrique. Plus les chaînes sont courtes, plus elles migrent loin et tous les fragments d'une même taille migrent à la même distance. On obtient alors une succession de bandes colorées, chacune correspondant au dernier nucléotide incorporé. Il suffit alors de lire la succession des couleurs pour connaître l'ordre des nucléotides, c'est-à-dire la séquence de l'ADN, étape assurée automatiquement par les détecteurs du séquenceur.

Transmission :
L’allèle de cette maladie étant dominant, il suffit d’être hétérozygote pour hériter de la maladie, une personne malade a donc un risque sur deux de transmettre la maladie à ses enfants. Dans 50% des cas, il est aussi possible qu’un enfant malade soit né de deux parents indemnes car les néo-mutations sont très fréquentes. Dans de très rares cas, il est possible qu’un individu soit atteint de mosaïque germinale, c'est-à-dire qu’il est totalement sain mais qu’il possède des gamètes mutés. Les gonades possèdent alors une double population cellulaire, des cellules mutées et des cellules saines ; il peut donc ou non transmettre la maladie à certains de ses enfants.


Vivre avec :
Voici un article qui traite de ce sujet
http://kokcinelo.fr/wp-content/uploads/2011/07/ML-13-JUIN.png

La recherche :
En 2012, les médecins décident de tester un nouveau traitement pour guérir la neurofibromatose. Leur but est de prouver que la molécule nommée évérolimus peut réduire la taille des nodules mettant en danger la vie des patients qui n'on pas de solution chirurgicale, dans un second temps ils recherchent si cette molécule peut aussi avoir un effet sur les neurofibromes cutanés. Il est mis à disposition du personnel médical un tutoriel afin de diagnostiquer les premiers symptômes de la maladie plus précocément. Ils cherchent à obtenir plus d'informations au niveau moléculaire et cellulaire du fonctionnement du gène NF1. Des recherches sont effectuées afin de trouver la mutation des mélanocytes, et afin de trouver un traitement à d’autres anomalies. Le gène défaillant est entièrement étudier afin de trouver la mutation. Des expériences sont menées sur des femmes en âge de procréer et/ou qui prennent une contraception orale afin de déterminer le rôle des hormones stéroïdiennes, ou non, dans l’apparition et le développement des neurofibromes afin de pouvoir proposer un diagnostic anténatal. Des études sont menées sur la minéralisation osseuse afin de définir un type de personne à risques et de leurs proposer une stratégie thérapeutique pour limiter les risques de fractures. Enfin le projet vise à améliorer la prise en charge des troubles neuropsychologiques ainsi que leurs impacts. On espère également améliorer le secteur de connaissance de la maladie en vue d’une prise en charge plus rapide et plus efficace.

Traitement évérolimus:
Evérolimus, C53H83NO14, est une molécule analogue de la molécule sirolimus, C51H79NO13, utilisée pour soigner des maladies semblables à la neurofibromatose. Cette molécule est utilisée chez des personnes ayant de gros neurofibromes internes, sans solution chirurgicale. Le but est de réduire d'au moins 30% la masse tumorale interne afin d’éviter que les nodules ne deviennent visibles. Ce traitement est en expérimentation depuis 2011. Il s'étend sur une durée d'un an durant laquelle les patients doivent prendre leur traitement tous les jours, faire des analyses sanguines et des IRM tous les mois. L'issue du traitement est incertaine mais c'est actuellement le meilleur espoir pour les malades.

Résultats :
Le premier traitement a été testé sur 9 patients, dont deux âgés de moins de 18 ans. Sur les 9 patients, 3 ont dû arrêter pour cause d’apparition de tumeurs, 3 autres pour causes d’un manque de réponses positives et un dernier à cause d’un développement d’une pneumonie et de difficultés respiratoires. Uniquement 2 des 9 patients ont compléter les douze tests requis pour l’étude du traitement. Malgré cela, 3 des 9 patients ont vue une diminution volumétrique des tumeurs au bout du 8 mois de traitement, en moyenne.
Mutation :
Le gène NF1 étant très grand il est trop compliqué pour les chercheurs de le séquencer. Cette difficulté s’ajoute au fait que les mutations ne sont que très rarement identique d’un individu à l’autre, ce qui empêche de cibler la mutation type. Ces mutations peuvent être soit une délétion ce qui décale la lecture des codons et provoque un codon stop anticipé, ainsi la protéine est plus petite, c’est le début de la maladie. Parfois le gène NF1 n’est pas muté, la mutation se retrouve alors sur le gène speed one qui est à l’origine du syndrome de Legius.
Les anomalies génétiques possibles sont très nombreuses et difficiles à mettre en évidence. Chacune des méthodes disponibles actuellement ne permet de détecter qu’un certain pourcentage d’anomalies, ce qui oblige les laboratoires à coupler plusieurs techniques pour accroître le taux de détection. Cette recherche directe est rarement effectuée aujourd'hui. Le taux de détection des mutations est d’un peu moins de 90%.
Conclusion :
La neurofibromatose est une maladie génétique, héréditaire. Mais elle peut aussi être provoquée par néo-mutation. Celle-ci se manifeste par des tâches pigmentées (macules café au lait), des tâches de rousseur aux aisselles et des neurofibromes de taille variée. Certaines des personnes atteintes présentent des troubles neurologiques et des anomalies musculosqueletiques. Dans 2 à 5% des cas, il se produit une dégénérescence maligne des neurofibromes.
La mutation qui en est sa cause est très difficile à trouver et très souvent différente d’une personne à l’autre. Il est donc difficile de prévoir la maladie bien que l’amélioration du séquençage augmente les chances de trouver une mutation. De nouvelles techniques sont mises en place afin de diagnostiquer plus tôt la maladie et d’avoir une meilleure prise en charge des patients. Un traitement est actuellement en expérimentation, il a pour but de réduire la taille des nodules douloureux qui n’ont pas de solution chirurgicale. Cependant la vie des malades reste difficile notamment à cause du regard des autres mais aussi à cause des atteintes cérébrales qui provoquent des écarts avec les personnes non malades, surtout chez les enfants.
Actuellement il y a des associations comme "Kokcinelo" ou encore "Neurofibromatoses" qui ont été créées pour soutenir, accompagner, conseiller ces personnes. On peut s'étonner que le Téléthon ne reverse rien pour la neurofibromatose qui est une maladie génétique rare (article). Il est tout de même possible de faire des dons pour aider la recherche. Effectivement les chercheurs ont découvert une nouvelle molécule « évérolimus ». malheureusement la recherche n’évolue pas asser rapidement. Les malades doivent cependant garder l'espoir qu’un jour la médecine sera en mesure de guérir la neurofibromatose.
C'est une affection génétique, longtemps confondue avec la "maladie d'éléphant man". Elle atteint 15 000 personnes en France.
Les maladies génétiques :
Les maladies génétiques désignent l'ensemble des maladies qui sont causées par un ou plusieurs gènes défectueux ou par une anomalie chromosomique. Elles peuvent être héréditaires ou non. Ainsi, les maladies géniques qui concernent des gènes défectueux sont transmissibles à la descendance. À la différence des maladies chromosomiques qui ne sont pas héréditaires. La NF1 fait partie des maladies génétiques rares les plus connues, en France elle touche une personne sur 3000 à 4000.
Le blog que nous avons créé vous informe sur cette maladie rare et vous dirige sur le site de l'association Kokcinelo
Le chromozome 17 :
Le gène responsable de cette maladie est situé sur le bras long du chromozome 17. Il fabrique une protéine , la neurofibromine, responsable de la maladie. La neurofibromatose est une maladie plus ou moins sévère (de type 1 ou 2) avec une évolution imprévisible ce qui empêche d'effectuer un pronostic précis.


Les symptômes :
Les symptômes sont présents dès le plus jeune âge, on observe :
► des marques brunes claires sur la peau, appelées taches « café au lait » d'un diamètre supérieur à 1,5 cm après la puberté ou à 0,5 cm avant la puberté
► des taches de rousseur sous l'aine ou sous les bras
► des tumeurs non cancéreuses sous la peau (ou plus profondes dans le corps)
► des taches brunes sur l'iris, appelées nodules de Lisch
► des annomalies osseuses, comme les jambes arquées, une petite taille, ainsi qu'une scoliose
► des tumeurs sur le nerf optique (gliome)
Quelques symptômes en image :





Diagnostic :
Avant pour que les médecins puissent commencer à diagnostiquer la maladie, il fallait que le patient présente au moins six tâches café au lait d’un diamètre supérieur à 1.5 cm sur la totalité du corps. Après la confirmation de ce symptôme, on pratiquait une IRM afin d’observer des bright spots dans le cerveau. Ensuite on procédait à une observation du profil de l’œil à l’aide d’une lampe à fente à la recherche d'hamartome iridien.
Si tous ces symptômes étaient réunis on réalisait un séquençage du gène NF1, et dans 80% à 90% des cas le gène présentait une mutation.
Actuellement des techniques permettent d’améliorer le séquençage des gènes, on procède donc au séquençage après le premier diagnostic. Chez les enfants de moins de cinq ans, des visites régulières chez un ophtalmologiste sont obligatoires afin de détecter un possible gliome du nerf optique. Les adultes quant à eux s’auto-palpent afin de dépister eux même le gliome.
 |
| Cancer intracranien |
Complications :
En cas de complications les tumeurs peuvent devenir malignes, il se développe également des tumeurs cérébrales, des tumeurs des glandes surrénales, des tumeurs localisées en bas des reins. La première peut entraîner des déficiences mentales, des crises d'épilépsies et des troubles de l'apprentissage et du comportement. On différencie les tumeurs cérébrales des patients atteints de la NF1 aux tumeurs des autres patients qui nécessitent des thérapies différentes.
Les tumeurs des glandes surrénales entrainent une augmentation du taux d'adrénaline dans le sang et donc de l'hypertension.
Ces complications sont imprévisibles d'ou la difficulté de donner un pronostic au patient.
Dépistage :
Il est possible de dépister la maladie dans deux cas :
Lors d'un diagnostic prénatal ou préimplantatoir ou lorsque l'on envisage un diagnostic précoce chez l’enfant présentant des manifestations incomplètes. Ces techniques sont pratiquées dans trois centres en France: le CHR d'Orléans, Hôpital Antoine-Béclère, Hôpital Necker.
 | |
| Extraction d'une cellule oeuf, obtenue par fécondation in vitro qui sera analysée en laboratoire |
Le séquençage :
Le séquençage de l'ADN est une technique qui a révolutionné la biologie moléculaire dans les années 1970. L'ADN contenu dans chacune de nos cellules, contient l'information nécessaire à l'expression des gènes.
La méthode de Sanger, qui repose sur la biologie moléculaire, est désormais la plus utilisée. Bien que le séquençage ait beaucoup évolué et soit désormais automatisé, il repose généralement sur l'utilisation de composants biologiques qui existent naturellement dans les cellules.
Pour le séquençage, tout se passe initialement dans un tube, en présence de l'ADN à séquencer, des nucléotides normaux et des nucléotides modifiés, d'une amorce et de l'ADN polymérase.
L'ADN polymérase utilise aléatoirement les nucléotides présents dans le tube pour copier le brien mère en synthétisant un ADN de séquence complémentaire, du moment que la base (A, C, G ou T) est respectée, en respectant la complémentaritédes bases A-T et C-G.
Lorsque l'ADN polymérase choisit par hasard un nucléotide modifié (didésoxynucléotide, sans groupement -OH) pour synthétiser le brien d'ADN complémentaire, la synthèse s'interrompt prématurément. Chaque didésoxynucléotide étant associé à un fluorochrome différent (A vert, T rouge, C bleu et G jaune) une chaîne qui se termine par exemple par un A sera verte.
Puisqu'un grand nombre de réactions de synthèse ont lieu dans le tube, il existe des fragements de différentes tailles (correspondant à un arrêt de la synthèse à chaque didésoxynucléotide) et de nombreuses copies d'une même taille. Ces chaînes commencent toutes au même endroit sur l'ADN à séquencer (déterminé par la séquence de l'amorce utilisée), toutes celles qui possèdent la même longueur se terminent donc par le même didésoxynucléotide marqué.
Il est alors possible de séparer les chaînes d'ADN obtenues en fonction de leurs tailles, sur un gel d'acrylamide en présence d'un courant électrique. Plus les chaînes sont courtes, plus elles migrent loin et tous les fragments d'une même taille migrent à la même distance. On obtient alors une succession de bandes colorées, chacune correspondant au dernier nucléotide incorporé. Il suffit alors de lire la succession des couleurs pour connaître l'ordre des nucléotides, c'est-à-dire la séquence de l'ADN, étape assurée automatiquement par les détecteurs du séquenceur.

Transmission :
L’allèle de cette maladie étant dominant, il suffit d’être hétérozygote pour hériter de la maladie, une personne malade a donc un risque sur deux de transmettre la maladie à ses enfants. Dans 50% des cas, il est aussi possible qu’un enfant malade soit né de deux parents indemnes car les néo-mutations sont très fréquentes. Dans de très rares cas, il est possible qu’un individu soit atteint de mosaïque germinale, c'est-à-dire qu’il est totalement sain mais qu’il possède des gamètes mutés. Les gonades possèdent alors une double population cellulaire, des cellules mutées et des cellules saines ; il peut donc ou non transmettre la maladie à certains de ses enfants.

Vivre avec :
Le plus souvent la neurofibromatose 1
est compatible avec une vie normale sauf en cas de complications graves.
Chez certains enfants les troubles de l’apprentissage peuvent retentir
sur la scolarité et nécessiter une prise en charge spécifique pour
éviter qu’un écart se creuse avec le reste de la classe.
A l’âge adulte, les personnes atteintes
sont habituellement bien insérées au niveau social et professionnel. Les
neurofibromes cutanés peuvent néanmoins avoir un retentissement assez
important sur la qualité de vie.
http://kokcinelo.fr/wp-content/uploads/2011/07/ML-13-JUIN.png
{kind=link}
La recherche :
En 2012, les médecins décident de tester un nouveau traitement pour guérir la neurofibromatose. Leur but est de prouver que la molécule nommée évérolimus peut réduire la taille des nodules mettant en danger la vie des patients qui n'on pas de solution chirurgicale, dans un second temps ils recherchent si cette molécule peut aussi avoir un effet sur les neurofibromes cutanés. Il est mis à disposition du personnel médical un tutoriel afin de diagnostiquer les premiers symptômes de la maladie plus précocément. Ils cherchent à obtenir plus d'informations au niveau moléculaire et cellulaire du fonctionnement du gène NF1. Des recherches sont effectuées afin de trouver la mutation des mélanocytes, et afin de trouver un traitement à d’autres anomalies. Le gène défaillant est entièrement étudier afin de trouver la mutation. Des expériences sont menées sur des femmes en âge de procréer et/ou qui prennent une contraception orale afin de déterminer le rôle des hormones stéroïdiennes, ou non, dans l’apparition et le développement des neurofibromes afin de pouvoir proposer un diagnostic anténatal. Des études sont menées sur la minéralisation osseuse afin de définir un type de personne à risques et de leurs proposer une stratégie thérapeutique pour limiter les risques de fractures. Enfin le projet vise à améliorer la prise en charge des troubles neuropsychologiques ainsi que leurs impacts. On espère également améliorer le secteur de connaissance de la maladie en vue d’une prise en charge plus rapide et plus efficace.

Traitement évérolimus:
Evérolimus, C53H83NO14, est une molécule analogue de la molécule sirolimus, C51H79NO13, utilisée pour soigner des maladies semblables à la neurofibromatose. Cette molécule est utilisée chez des personnes ayant de gros neurofibromes internes, sans solution chirurgicale. Le but est de réduire d'au moins 30% la masse tumorale interne afin d’éviter que les nodules ne deviennent visibles. Ce traitement est en expérimentation depuis 2011. Il s'étend sur une durée d'un an durant laquelle les patients doivent prendre leur traitement tous les jours, faire des analyses sanguines et des IRM tous les mois. L'issue du traitement est incertaine mais c'est actuellement le meilleur espoir pour les malades.
Résultats :
Le premier traitement a été testé sur 9 patients, dont deux âgés de moins de 18 ans. Sur les 9 patients, 3 ont dû arrêter pour cause d’apparition de tumeurs, 3 autres pour causes d’un manque de réponses positives et un dernier à cause d’un développement d’une pneumonie et de difficultés respiratoires. Uniquement 2 des 9 patients ont compléter les douze tests requis pour l’étude du traitement. Malgré cela, 3 des 9 patients ont vue une diminution volumétrique des tumeurs au bout du 8 mois de traitement, en moyenne.
Mutation :
Le gène NF1 étant très grand il est trop compliqué pour les chercheurs de le séquencer. Cette difficulté s’ajoute au fait que les mutations ne sont que très rarement identique d’un individu à l’autre, ce qui empêche de cibler la mutation type. Ces mutations peuvent être soit une délétion ce qui décale la lecture des codons et provoque un codon stop anticipé, ainsi la protéine est plus petite, c’est le début de la maladie. Parfois le gène NF1 n’est pas muté, la mutation se retrouve alors sur le gène speed one qui est à l’origine du syndrome de Legius.
Les anomalies génétiques possibles sont très nombreuses et difficiles à mettre en évidence. Chacune des méthodes disponibles actuellement ne permet de détecter qu’un certain pourcentage d’anomalies, ce qui oblige les laboratoires à coupler plusieurs techniques pour accroître le taux de détection. Cette recherche directe est rarement effectuée aujourd'hui. Le taux de détection des mutations est d’un peu moins de 90%.
Conclusion :
La neurofibromatose est une maladie génétique, héréditaire. Mais elle peut aussi être provoquée par néo-mutation. Celle-ci se manifeste par des tâches pigmentées (macules café au lait), des tâches de rousseur aux aisselles et des neurofibromes de taille variée. Certaines des personnes atteintes présentent des troubles neurologiques et des anomalies musculosqueletiques. Dans 2 à 5% des cas, il se produit une dégénérescence maligne des neurofibromes.
{kind=link}
La mutation qui en est sa cause est très difficile à trouver et très souvent différente d’une personne à l’autre. Il est donc difficile de prévoir la maladie bien que l’amélioration du séquençage augmente les chances de trouver une mutation. De nouvelles techniques sont mises en place afin de diagnostiquer plus tôt la maladie et d’avoir une meilleure prise en charge des patients. Un traitement est actuellement en expérimentation, il a pour but de réduire la taille des nodules douloureux qui n’ont pas de solution chirurgicale. Cependant la vie des malades reste difficile notamment à cause du regard des autres mais aussi à cause des atteintes cérébrales qui provoquent des écarts avec les personnes non malades, surtout chez les enfants.
Actuellement il y a des associations comme "Kokcinelo" ou encore "Neurofibromatoses" qui ont été créées pour soutenir, accompagner, conseiller ces personnes. On peut s'étonner que le Téléthon ne reverse rien pour la neurofibromatose qui est une maladie génétique rare (article). Il est tout de même possible de faire des dons pour aider la recherche. Effectivement les chercheurs ont découvert une nouvelle molécule « évérolimus ». malheureusement la recherche n’évolue pas asser rapidement. Les malades doivent cependant garder l'espoir qu’un jour la médecine sera en mesure de guérir la neurofibromatose.
{kind=link}
Je m'appelle Hoover, ma fille de 18 ans, Tricia a été diagnostiquée avec l'herpès il y a 3 ans. Depuis lors, nous sommes passés d'un hôpital à un autre. Nous avons essayé toutes sortes de pilules, mais tous les efforts pour se débarrasser du virus étaient vains. Les bulles ont continué à réapparaître après quelques mois. Ma fille utilisait des comprimés d'acyclovir à 200 mg. 2 comprimés toutes les 6 heures et 15 g de crème de fusitine. et H5 POT. Le permanganate avec de l'eau doit être appliqué deux fois par jour, mais tous ne donnent toujours pas de résultats. Donc, j'étais sur Internet il y a quelques mois, pour chercher d'autres moyens de sauver mon fils unique. Ce n'est qu'alors que j'ai rencontré un commentaire sur le traitement à base de plantes du Dr Imoloa et j'ai décidé de l'essayer. Je l'ai contacté et il a préparé des herbes et les a envoyées, ainsi que des conseils sur la façon de les utiliser via le service de messagerie DHL. ma fille l'a utilisé comme dirigé par le Dr Imoloa et en moins de 14 jours, ma fille a retrouvé la santé. Vous devez contacter dr imoloa aujourd'hui directement à son adresse e-mail pour tout type de problème de santé; lupus, ulcère de la bouche, cancer de la bouche, douleurs corporelles, fièvre, hépatite ABC, syphilis, diarrhée, VIH / sida, maladie de Huntington, acné au dos, insuffisance rénale chronique, maladie d'Addison, douleur chronique, douleur de Crohn, fibrose kystique, fibromyalgie, inflammatoire Maladie intestinale, mycose des ongles, maladie de Lyme, maladie de Celia, lymphome, dépression majeure, mélanome malin, manie, mélorhéostose, maladie de Ménière, mucopolysaccharidose, sclérose en plaques, dystrophie musculaire, polyarthrite rhumatoïde maladie d'Alzheimer, maladie de Parkinson, cancer du vagin, épilepsie , Maladie auto-immune, Douleurs dorsales, Entorse dorsale, Trouble bipolaire, Tumeur cérébrale, Maligne, Bruxisme, Boulimie, Disque cervical, Maladie cardiovasculaire, Tumeurs, Maladie respiratoire chronique, Trouble mental et comportemental, Fibrose kystique, Hypertension, Diabète, Asthme, Auto-immune arthrite des milieux inflammatoires ed. maladie rénale chronique, maladie articulaire inflammatoire, impuissance, spectre d'alcool feta, trouble dysthymique, eczéma, tuberculose, syndrome de fatigue chronique, constipation, maladie inflammatoire de l'intestin. et beaucoup plus; contactez-le à drimolaherbalmademedicine@gmail.com./ également avec whatssap- + 2347081986098.
RépondreSupprimerBonjour à tous, je suis Jesus Mckinney du Texas et je veux juste dire un très grand merci aux services de prêt financier de Benjamin Lee pour leur sincérité, ouverture, transparence, véracité, amour et soutien pendant et après avoir obtenu des fonds de prêt de leur part. J'ai traversé beaucoup de choses et le temps ne me permet pas de dire tout ce que j'ai vécu en ligne chez l'invité ou d'obtenir un prêt pour obtenir une maison ici aux États-Unis, mais Dieu a répondu à mes prières grâce au soutien et à l'amour de Benjamin Lee qui m'a embrassé et m'a compris malgré mon doute initial et mon manque de sérieux et avec son bon cœur et son amour, je suis maintenant propriétaire d'une maison grâce à ses fonds de prêt à taux d'intérêt de 2% et j'ai le vœu de répandre cette nouvelle et de dire au monde qu'il existe encore et peu de bonnes sociétés de prêt en ligne qui peuvent aider et raviver un os sec. ceci parce que j'ai prouvé et je jure devant Dieu dans le Ciel que cette histoire est réelle et aussi l'histoire de mon expérience avec eux.
RépondreSupprimerContactez-les dès aujourd'hui. Texte WhatsApp: (+1989-394-3740) Courriel: 247officedept@gmail.com